De getoonde personages zijn echte patiënten en de vereiste toestemming om hun verhalen te gebruiken is verkregen van de patiënten en families. Foto's zijn uitsluitend bedoeld ter illustratie.
WAT IS SPINALE
MUSCULAIRE ATROFIE (SMA)?
SMA is een zeldzame genetische neuromusculaire ziekte die het deel van het zenuwstelsel aantast dat vrijwillige spierbewegingen controleert.1
De getoonde personages zijn echte patiënten en de vereiste toestemming om hun verhalen te gebruiken is verkregen van de patiënten en families. Foto's zijn uitsluitend bedoeld ter illustratie.
WAT IS SPINALE MUSCULAIRE ATROFIE (SMA)?
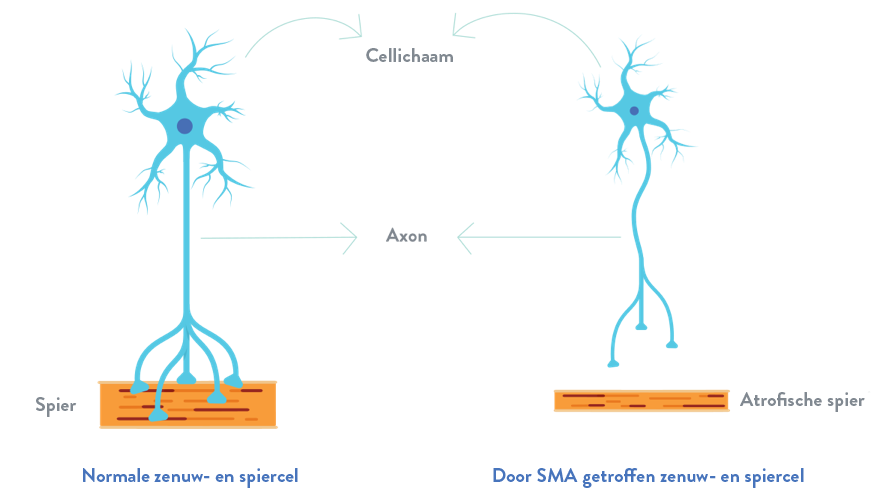
Bij spinale musculaire atrofie is er een verlies van belangrijke cellen in het ruggenmerg, namelijk motorneuronen, die essentieel zijn voor spierkracht en beweging. Deze motorneuronen regelen de spieractiviteit door signalen vanuit het centrale zenuwstelsel (CZS) te sturen. Dat is het deel van het zenuwstelsel dat de hersenen en het ruggenmerg omvat.1,2
Het verlies van de functionerende motorneuronen leidt tot progressieve spierzwakte en atrofie (de geleidelijke afname van massa en sterkte van de spieren), omdat spieren geen signalen meer krijgen vanuit het CZS.3
In tegenstelling tot vele andere zeldzame neuromusculaire ziekten begrijpen we goed wat de specifieke genetische oorzaak is van spinale musculaire atrofie.
WAT VEROORZAAKT SPINALE MUSCULAIRE ATROFIE?
Spinale musculaire atrofie wordt veroorzaakt door een mutatie in het survival motor neuron 1 (SMN1)-gen. Dit gen is verantwoordelijk voor de aanmaak van het survival motor neuron (SMN)-eiwit, dat instaat voor de gezondheid en de normale werking van motorneuronen.
Bij mensen met spinale musculaire atrofie zijn beide kopieën van het SMN1-gen gemuteerd, wat leidt tot een verminderde aanmaak van SMN-eiwit. Zonder een behoorlijke hoeveelheid SMN-eiwit sterven de motorneuronen in het ruggenmerg af, en zo krijgen de spieren niet de juiste signalen vanuit de hersenen.5
De degeneratie van motorneuronen leidt tot een geleidelijke afname in de
massa en sterkte van de spieren (atrofie).
Uitsluitend ter illustratie.
WAT BETEKENT SPINALE MUSCULAIRE ATROFIE VOOR EEN KIND?
Elk kind met spinale musculaire atrofie wordt anders getroffen, en men moet weten dat de symptomen sterk kunnen variëren naargelang van de aanvangsleeftijd en de ernst van de ziekte. Kinderen kunnen progressieve spierzwakte ondervinden in de spieren die het dichtst bij het midden van het lichaam liggen, zoals de schouders, dijen en het bekken. Deze spieren maken activiteiten mogelijk zoals kruipen, stappen, rechtop zitten en controleren van de hoofdbeweging. Ook ademhalen en slikken kunnen aangetast worden.7
SMA is een zeldzame, genetische neuromusculaire ziekte,2,5 en SMA is de belangrijkste genetische oorzaak van mortaliteit bij zuigelingen en peuters.1
Spinale musculaire atrofie heeft geen invloed op de neuronen die verantwoordelijk zijn voor cognitie, het mentale proces waarmee we kennis en inzicht opdoen door middel van gedachten, ervaring en de zintuigen.8,9 Volgens één studie hebben kinderen en adolescenten met spinale musculaire atrofie een normale intelligentie, met IQ-waarden binnen het standaardbereik.10
WAT MOET IK WETEN OVER SMN2?
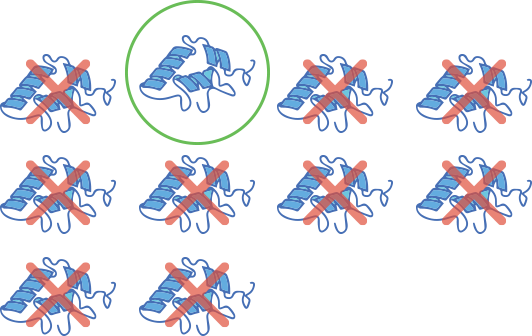
Alle personen met spinale musculaire atrofie hebben ten minste één “reservegen”, gekend als SMN2. Het SMN2-gen heeft een vergelijkbare structuur met dat van SMN1, maar slechts een kleine hoeveelheid (10%) van het SMN-eiwit dat het produceert, is volledig functioneel. Deze lage hoeveelheid SMN-eiwit is niet doeltreffend genoeg om de motorneuronen in het CZS te laten overleven.11-13
Het aantal SMN2-genen kan variëren. Een hoger aantal kopieën van SMN2 wordt geassocieerd met minder ernstige symptomen van spinale musculaire atrofie.5,13,14 De ziekte heeft veel verschillende symptomen en hoewel er een sterk verband is tussen het aantal SMN2-kopieën en de ernst van de ziekte, zijn er uitzonderingen. Deskundigen raden daarom aan zorgbeslissingen te nemen op basis van het functionele vermogen van het kind en niet op basis van het aantal kopieën van SMN2. Het aantal SMN2-kopieën wordt wel gebruikt als een hoofdcriterium voor registratie in klinische studies.2,15
Te wijten aan een tekort van het gen SMN1...1
Twee kopieën
Beide kopieën verwijderd of beschadigd
Geen SMN-eiwit aangemaakt
Gedeeltelijk gecompenseerd door het gen SMN2...1
Aantal kopieën is variabel
Slechts 10% SMN is functioneel
HOE WORDT SPINALE MUSCULAIRE ATROFIE OVERGEËRFD?
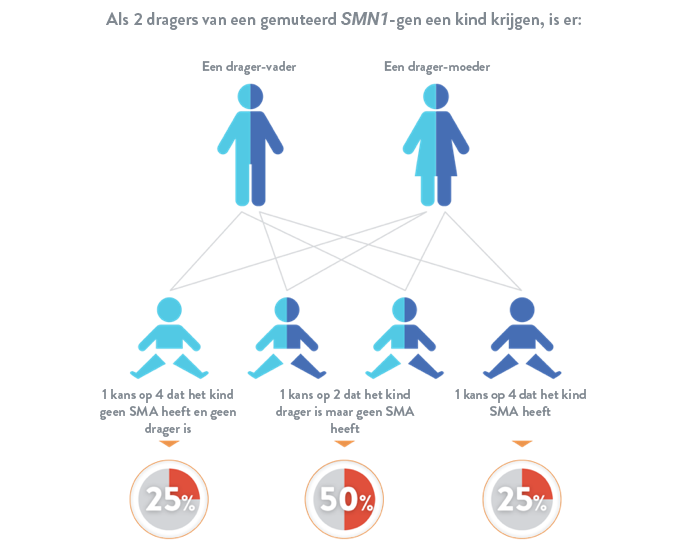
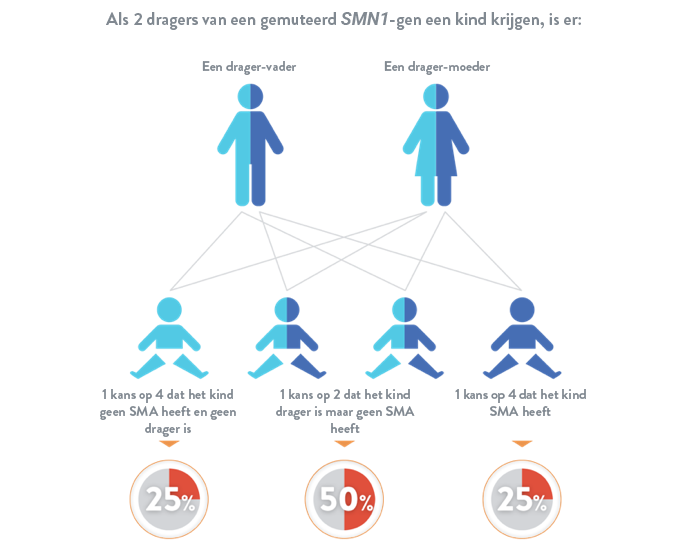
Spinale musculaire atrofie is een autosomaal recessieve ziekte, wat betekent dat een kind pas risico loopt als hij of zij van elke ouder 1 gemuteerd SMN1-gen erft. Als een kind slechts 1 gemuteerd SMN1-gen overerft, wordt het beschouwd als een "drager", maar heeft het meestal geen symptomen van spinale musculaire atrofie.16
Als spinale musculaire atrofie in uw familie voorkomt, is de kans meer dan gemiddeld dat u een drager bent. Wanneer u gaat beslissen of u kinderen wil, kan het nuttig zijn om uw arts te raadplegen om te weten te komen welke mutatie(s) er in uw familie vaak voorkomt/voorkomen. Zodra de mutatie(s) in uw familie gekend is/zijn, kan vastgesteld worden welke test voor uw situatie geschikt is:
Als de mutatie(s) in uw familie SMN1-deleties is/zijn, dan kan een test van het aantal kopieën voor u geschikt zijn
Als de mutatie(s) in uw familie een subtielere verandering in het gen omvat(ten), dan kunnen uw arts en het laboratorium beslissen of een test om die specifieke verandering op te sporen mogelijk is
Veel laboratoria en ziekenhuizen bieden een dragertest aan om te bepalen of 1 of beide ouders drager van het gemuteerde SMN1-gen is/zijn. Zo kunnen personen en gezinnen te weten komen of ze een risico lopen om een kind te krijgen met spinale musculaire atrofie.17 Een klinisch geneticus is opgeleid om informatie over genetische risico's, onderzoek en diagnose te verduidelijken naar gezinnen toe.
Kom hier te weten wat de rol van een klinisch geneticus inhoudt.
De getoonde personages zijn echte patiënten en de vereiste toestemming om hun verhalen te gebruiken is verkregen van de patiënten en families. Foto's zijn uitsluitend bedoeld ter illustratie.
Referenties
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
2.Mercuri E, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscl Disord. 2018;28(2):103-115.
6. Jones et al. Systematic review of incidence and prevalence of spinal muscular atrophy (SMA). European Journal of Paediatric Neurology. 2015;19(supp. 1):S64–S65.
7. Finkel R, et al. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7-9 November 2014, Heemskerk, Nederland. Neuromuscul Disord. 2015;25(7):593-602.
8. Qian Y., McGraw S., Henne J., Jarecki J., Hobby K., Yeh W.S. Understanding the experiences and needs of individuals with Spinal Muscular Atrophy and their parents: A qualitative study. BMC Neurol. 2015;15:1–12. doi: 10.1186/s12883-015-0473-3.
12. Cherry JJ, et al. Identification of novel compounds that increase SMN protein levels using an improved SMN2 reporter cell assay. J Biomol Screen. 2012;17(4):481-495.
13. Darras BT, Royden Jones H Jr, Ryan MM, De Vivo DC, eds. Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician’s Approach. 2nd Ed. Londen, VK: Elsevier; 2015.
14. Prior TW. Perspectives and diagnostic considerations in spinal muscular atrophy. Genet Med. 2010;12(3):145-152.
15. TREAT-NMD. Diagnostic testing and care of new SMA patients. Beschikbaar op: http://www.treat-nmd.eu/downloads/file/standardsofcare/sma/english/sma_soc_en.pdf. Geraadpleegd op 9 januari 2017.