Les symptômes peuvent comprendre une faiblesse musculaire progressive, un tonus musculaire faible et une fonte de la masse musculaire (atrophie). La faiblesse musculaire touche généralement les deux côtés du corps.1
Les personnages présentés sont de vrais patients et le consentement requis pour utiliser leurs histoires a été obtenu auprès des patients et de leurs familles. Les photographies sont à titre d'illustration uniquement.
Signes et symptômes
Partager la page
Chaque enfant peut ressentir différemment les symptômes, et la maladie est divisée selon différents types, en fonction de l’âge d’apparition de la maladie et en fonction de la capacité fonctionnelle. Il existe également des degrés de gravité dans chaque type, et jusqu’à 25 % des individus peuvent ne pas avoir de type précis.3
Caractéristiques de l’amyotrophie spinale
Cliquez sur les onglets afin d'obtenir les détails supplémentaires sur chaque type de SMA.
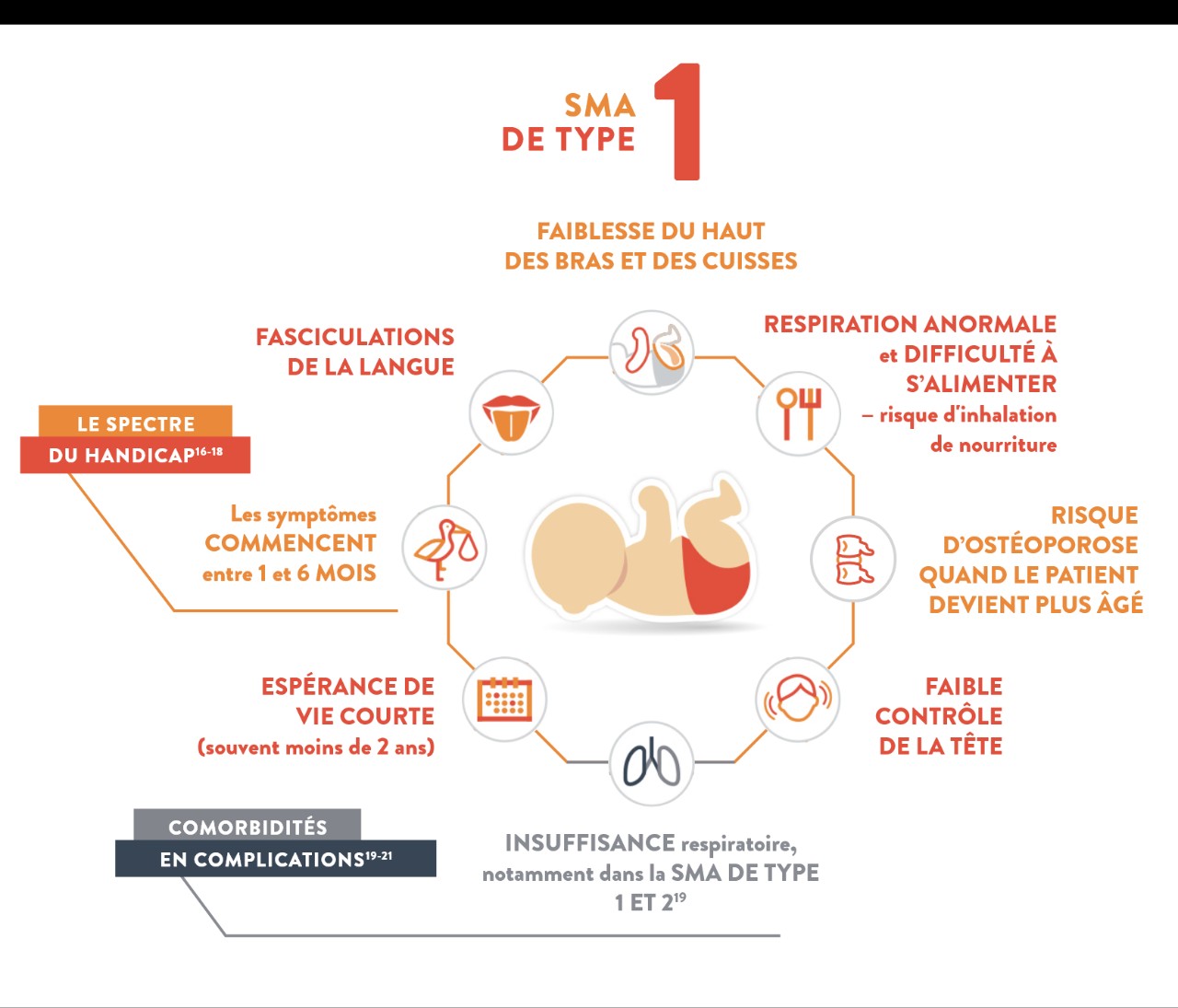
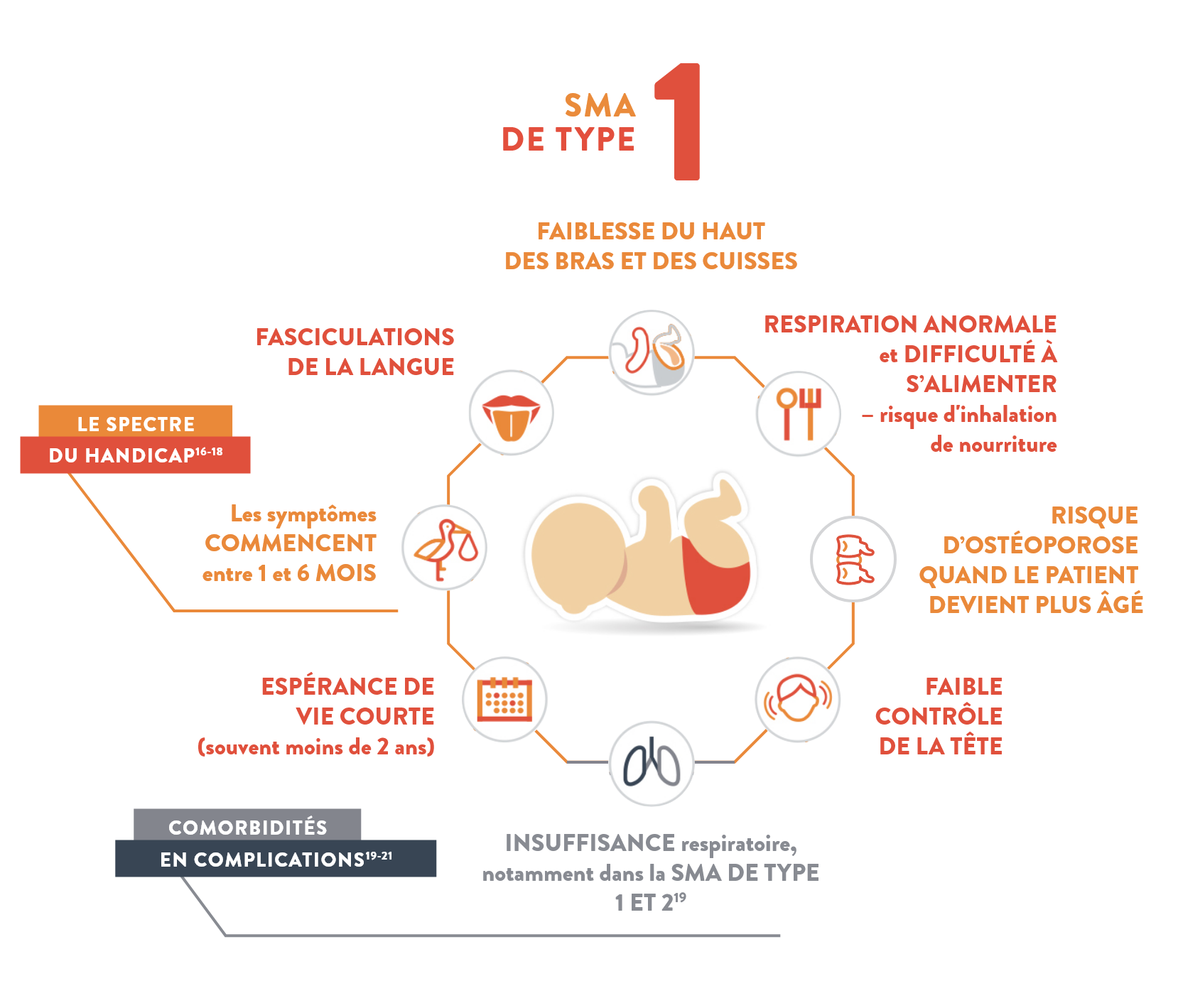
POSITION ASSISE IMPOSSIBLE
(« patients incapables de s’asseoir »)
Espérance de vie*
≤ 2 ANS4
* Si le patient n’est pas traité avec un traitement modifiant l’évolution de la maladie
Type
Type I
(également appelé maladie de Werdnig-Hoffmann)
Caractéristiques1,4,5-7
Faible contrôle de la tête
Toux faible
Larmoiement faible
Affaiblissement progressif des muscles sollicités par la mastication et la déglutition
Faible tonus musculaire
« Position de la grenouille » en position allongée
Faiblesse musculaire sévère des deux côtés du corps
Faiblesse progressive des muscles qui aident à respirer (muscles intercostaux), entraînant la forme caractéristique « en cloche » du thorax
Risque d'osthéoporose quand le patient devient plus âgé
Podijelite
Progrès moteur le plus avancé
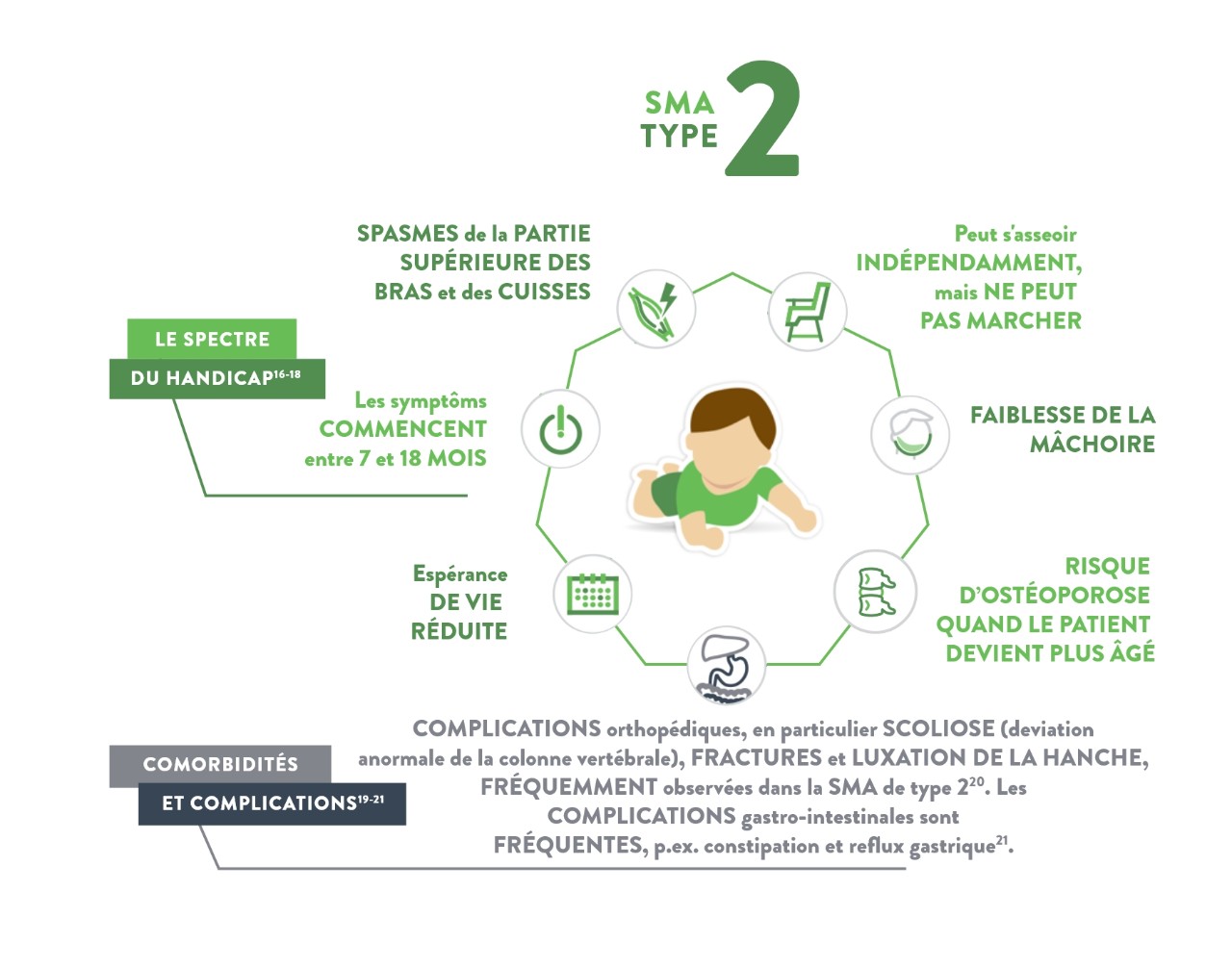
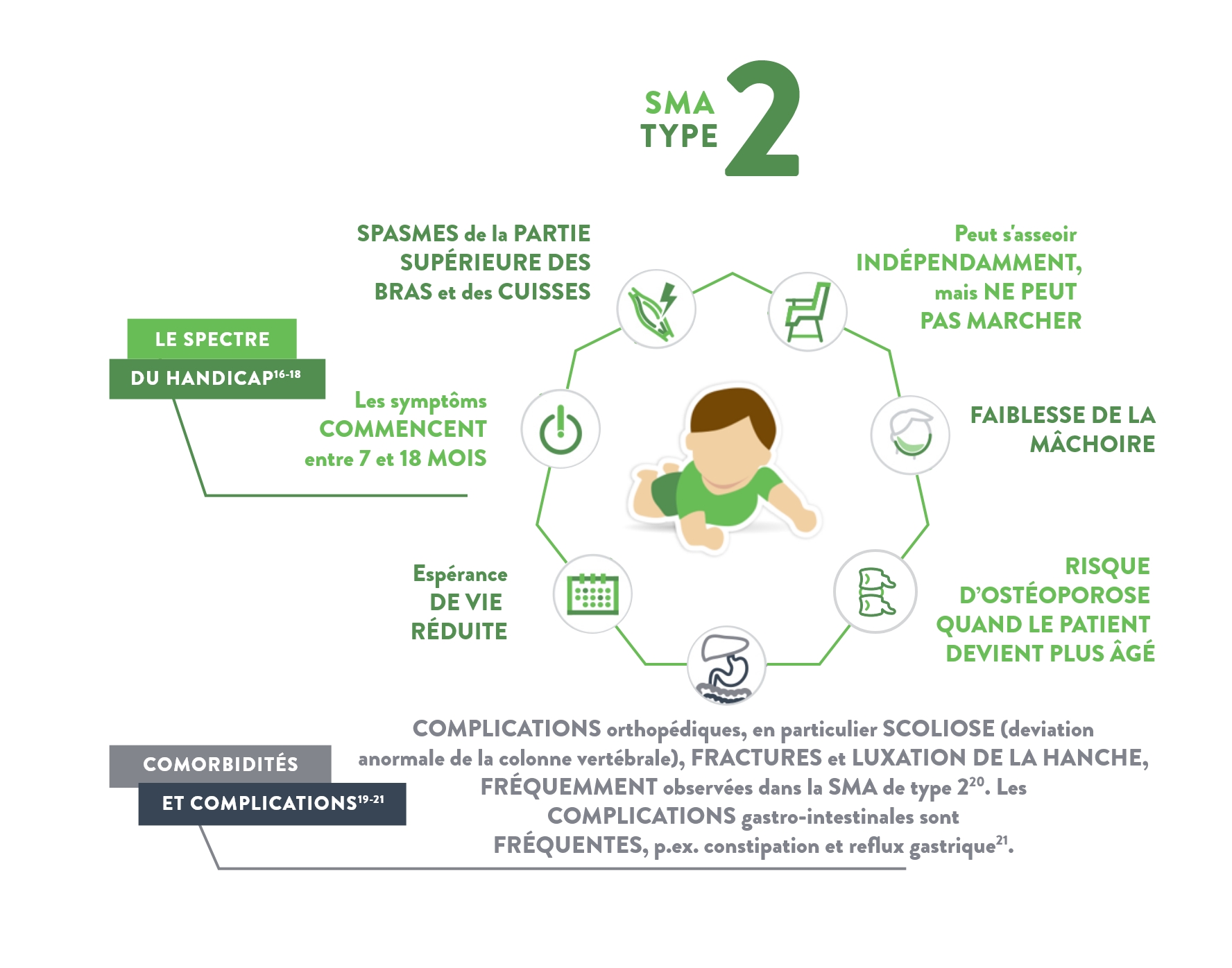
POSITION ASSISE SANS SOUTIEN
(« patients capables de s’asseoir »)
Espérance de vie*
> 2 ANS
70 % D’ENTRE EUX SONT ENCORE VIVANTS À L’ÂGE DE 25 ANS
* Si le patient n’est pas traité avec un traitement modifiant l’évolution de la maladie
Type
Type II
(également appelé syndrome de Dubowitz)
Caractéristiques1,5
Faiblesse musculaire
Les problèmes de déglutition, de toux et de respiration sont fréquents lorsque ces patients vieillissent
Symptômes de douleurs musculaires et de raideurs articulaires
La plupart des enfants atteints du type 2 développent des problèmes vertébraux tels qu’une scoliose (déviation de la colonne vertébrale), qui peut nécessiter une brassière de renfort ou une intervention chirurgicale
Risque d'osthéoporose quand le patient devient plus âgé
Podijelite
Progrès moteur le plus avancé
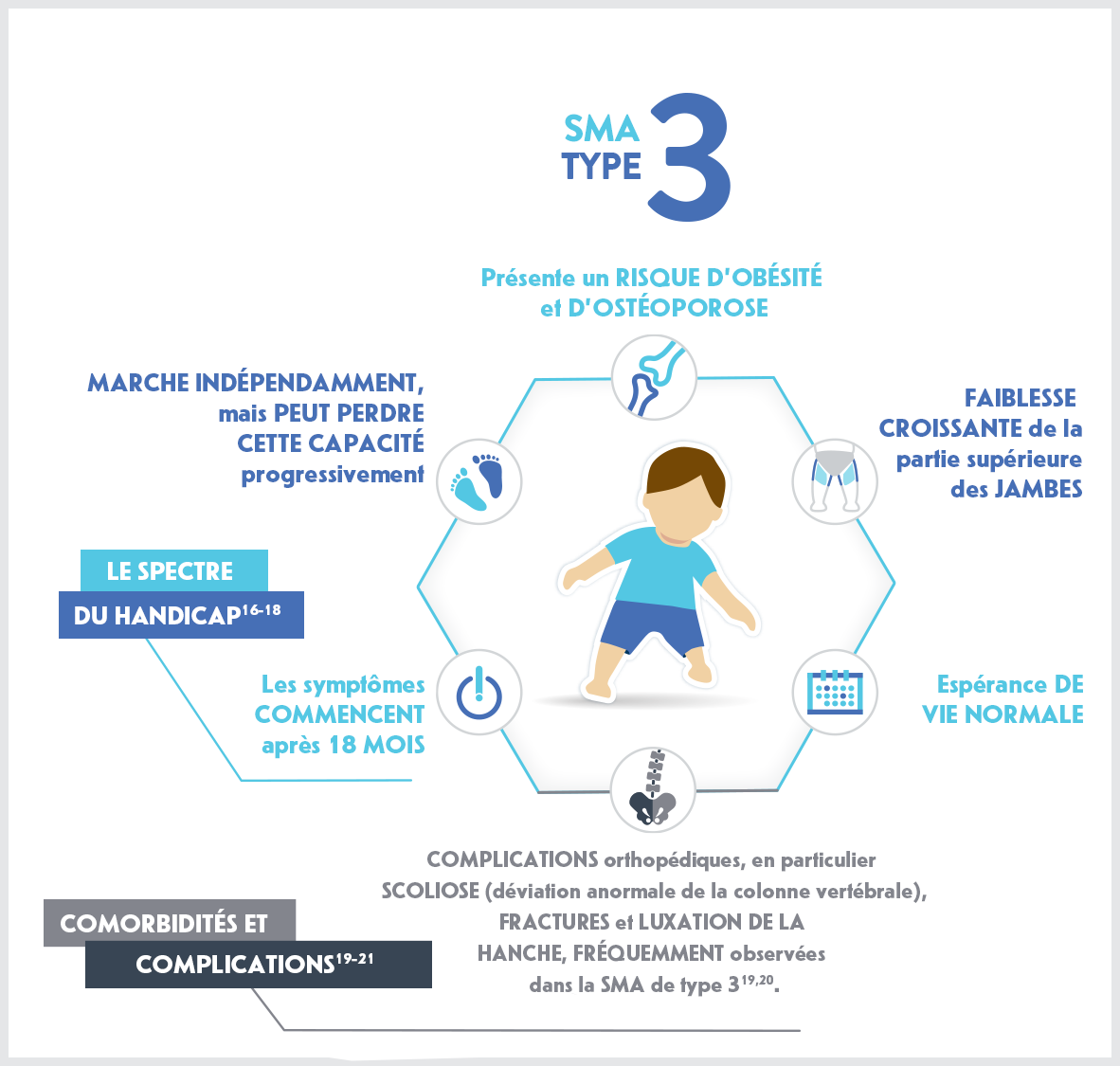
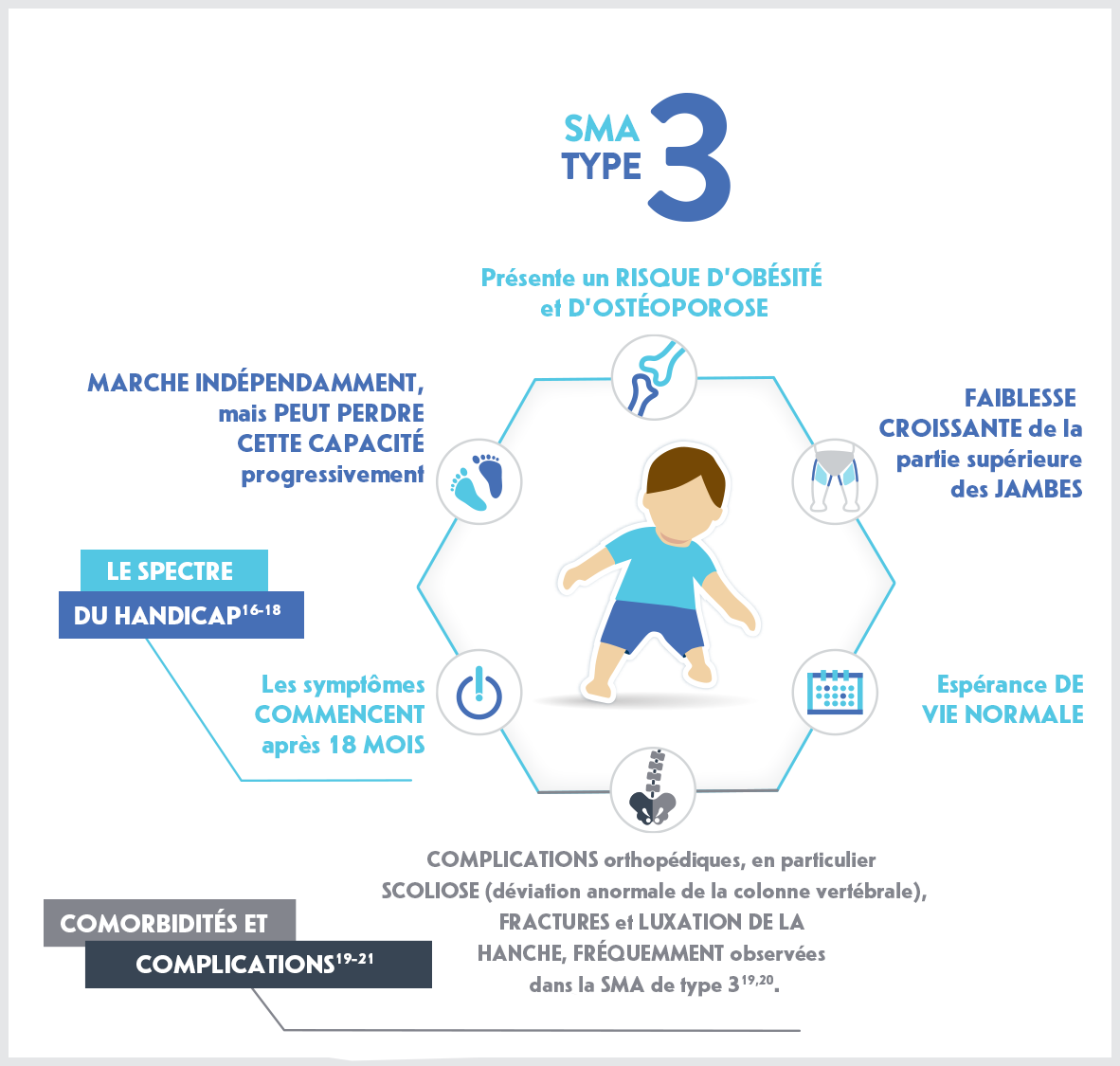
CAPABLE DE MARCHER SEUL(E)
(« marcheurs », même s’ils peuvent progressivement perdre cette capacité)
Espérance de vie*
NORMALE
* Si le patient n’est pas traité avec un traitement modifiant l’évolution de la maladie
Type
Type III
(également appelé maladie de Kugelberg-Welander)
Caractéristiques1,5
Scoliose
Problèmes de déglutition et de mastication
Les muscles des jambes sont généralement plus sévèrement affectés que les bras
Douleur musculaire
Symptômes de surmenage articulaire
Risque d'osthéoporose quand le patient devient plus âgé
Podijelite
Progrès moteur le plus avancé
TOUT
Espérance de vie*
NORMALE
* Si le patient n’est pas traité avec un traitement modifiant l’évolution de la maladie
Type
Type IV
Caractéristiques1,5
Les symptômes physiques sont similaires à ceux de l’amyotrophie spinale juvénile, avec l’apparition progressive de faiblesse, de tremblements et de contractions musculaires et sont initialement observés vers la fin de l’adolescence ou à l’âge adulte
Podijelite
L’amyotrophie spinale est souvent suspectée par un parent qui peut remarquer que son enfant n’atteint pas certains stades du développement moteur
Les parents peuvent remarquer que leur enfant n’atteint pas les étapes motrices typiques de son âge, comme la capacité à tenir sa tête droite, à faire une roulade ou à s’asseoir seul. La déglutition ou l’alimentation peuvent également devenir difficiles, et les enfants peuvent perdre la capacité à déglutir en toute sécurité sans s’étouffer ni inhaler les aliments dans les poumons (aspiration).9,13
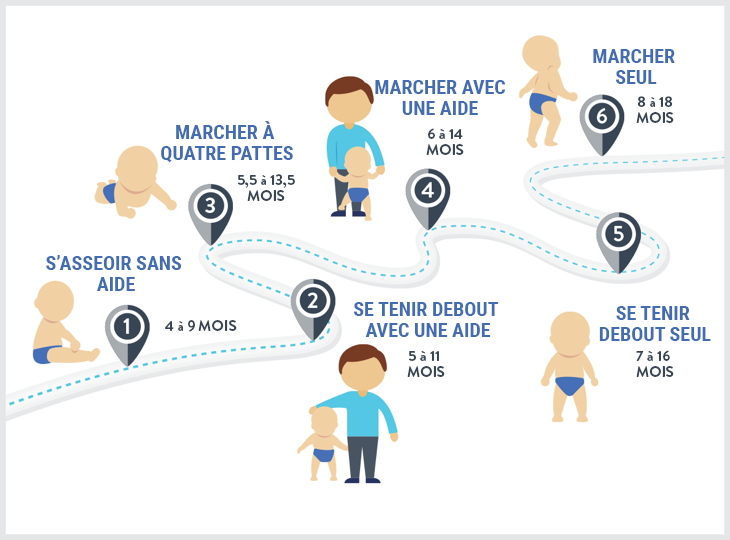
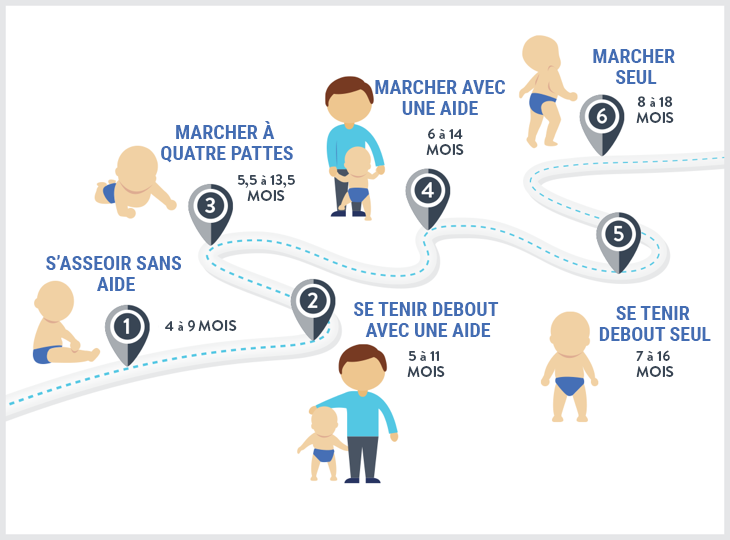
Alors que tous les bébés se développent à leur propre rythme, l’Organisation Mondiale de la Santé (OMS) propose les directives générales suivantes concernant les étapes motrices, dans le cadre de l’Étude multicentrique sur la référence de croissance (Multicentre Growth Reference Study [MGRS]) :14
Progrès moteurs dans la MGRS
STADE MOTEUR ATTEINT
CRITÈRES DE PERFORMANCE
S’asseoir sans aide
L’enfant s’assied droit avec la tête droite pendant plus de 10 secondes. N’utilise pas les bras ou les mains pour équilibrer le corps ou assurer la position assise.
Rampe sur les mains et les genoux
L’enfant se déplace vers l’avant ou vers l’arrière sur les mains et sur les genoux. Le ventre ne touche pas la surface de soutien. Mouvements continus et consécutifs, ≥ 3 d’affilée.
Se met debout sans assistance
L’enfant se tient debout sur les deux pieds, en se tenant à un support stable, comme un meuble. L’enfant se tient debout pendant ≥ 10 secondes.
Marche avec une assistance
L’enfant est en position verticale, le dos droit. Fait des mouvements latéraux ou vers l’avant, en se tenant à un support stable avec une main ou les deux. Une jambe avance tandis que l’autre soutient le poids du corps. L’enfant fait ≥ 5 pas.
Se met debout seul
L’enfant tient debout sur les deux pieds (mais pas sur la pointe des pieds), le dos droit. Les jambes soutiennent 100 % du poids, sans se tenir pendant ≥ 10 secondes.
Marche seul
L’enfant fait ≥ 5 pas seul, avec le dos droit. Une jambe avance tandis que l’autre soutient la majorité du poids. Il n’y a aucun contact avec une personne ou un objet.
Adapté de l’Étude multicentrique sur la référence de croissance établie par l’OMS.14
ÂGE DE L’ENFANT
MOUVEMENTS / ÉTAPES PHYSIQUES TYPIQUES
2 mois
Peut tenir la tête vers le haut et commencer à pousser lorsqu’il est couché sur le ventre
Effectue des mouvements plus doux avec les bras et les jambes
4 mois
Maintient la tête stable, sans aide
Pousse sur ses jambes lorsque ses pieds sont sur une surface dure
Peut être capable de rouler du ventre sur le dos
Peut tenir un jouet et le frapper contre des jouets suspendus
Porte ses mains à sa bouche
Se soulève pour reposer sur ses coudes lorsqu’il est sur le ventre
6 mois
Roule dans les deux sens (de l’avant vers l’arrière et de l’arrière vers l’avant)
Commence à s’asseoir sans aide
En position debout, ses jambes le supportent et il pourrait faire bondir son corps
Se balance vers l’avant et vers l’arrière, et rampe parfois vers l’arrière avant d’avancer
9 mois
Se tient debout en se tenant
Peut se mettre en position assise
S’assied sans aide
Prend appui pour se mettre debout
Rampe
1 an
Passe en position assise sans aide
Prend appui pour se mettre debout, marche en se tenant aux meubles
Peut faire quelques pas sans se tenir
Peut se tenir debout seul
Adapté selon la la liste de contrôle des étapes établie par les Centres américains pour le contrôle et la prévention des maladies (Centers for Disease Control and Prevention).13
Les rapports des parents concernant le développement moteur global de leur enfant sont généralement fiables. Le fait de communiquer au médecin des observations sur les retards moteurs possibles peut aider à établir une stratégie appropriée.2,15
Les personnages présentés sont de vrais patients et le consentement requis pour utiliser leurs histoires a été obtenu auprès des patients et de leurs familles. Les photographies sont à titre d'illustration uniquement.
Références
1.Mercuri E, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscl Disord. 2018;28(2):103-115.
2.Noritz GH, Murphy NA; and Neuromuscular Screening Expert Panel. Motor delays: early identification and evaluation. Pediatrics. 2013;131(6):e2016-e2027.
3.Kolb SJ, Kissel JT. Spinal muscular atrophy. Arch Neurol. 2011;68(8):979-984.
4.Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol. 2012;46(1):1-12.
5.Prior TW, Russman BS. Spinal muscular atrophy. NCBI Bookshelf Website. Disponible sur : http://www.ncbi.nlm.nih.gov/books/NBK1352/. Mise à jour le 14 novembre 2013. Consulté le 15 avril 2016.
6.Iannaccone ST. Modern management of spinal muscular atrophy. J Child Neurol. 2007;22(8):974-978.
7.Oskoui M, Levy G, Garland CJ, et al. The changing natural history of spinal muscular atrophy type 1. Neurology. 2007;69(20):1931-1936.
8.Darras BT, Royden Jones H Jr, Ryan MM, De Vivo DC, eds. Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician’s Approach. 2nd Ed. London, UK: Elsevier; 2015.
10.Online Mendelian Inheritance in Man. Neuronopathy, distal hereditary motor, type VA; HMN5A. http://www.omim.org/entry/600794. Modifié le 2 janvier 2014. Consulté le 22 avril 2016.
11.National Organization for Rare Diseases. Spinal muscular atrophy. https://rarediseases.org/rare-diseases/spinal-muscular-atrophy/. Mise à jour 2012. Consulté le 17 avril 2016.
12.Barkhaus PE et al. Kennedy Disease. Disponible sur : http://emedicine.medscape.com/article/1172604-overview. Mise à jour : 8 juin 2016. Consulté le 9 janvier 2017.
13.Centers for Disease Control and Prevention. Developmental milestones. Disponible sur : http://www.cdc.gov/ncbddd/actearly/milestones/. Mis à jour le 21 janvier 2016. Consulté le 27 avril 2016.
14.Wijnhoven TMA, de Onis M, Onyango AE, et al; for the WHO Multicentre Growth Reference Study Group. Assessment of gross motor development in the WHO Multricentre Growth Reference Study. Food Nutr Bull. 2004;25(1 suppl 1):S37-S45.
15.Lawton S, Hickerton C, Archibald AD, McClaren BJ, Metcalfe SA. A mixed methods exploration of families’ experiences of the diagnosis of childhood spinal muscular atrophy. Eur J Hum Genet. 2015;23(5):575-580.
16.Kolb SJ, Kissel JT. Neurol Clin.. 2015;33:831-46.
17.D'Amico A, et al. Orphanet J Rare Dis. 2011;6:71.
18.Lunn MR, Wang CH. Lancet. 2008;371:2120-33.
19.Haaker G, Fujak A. Appl Clin Genet. 2013;6:113-20.
20.Darras BT. Paediatr Clin North Am. 2015;62:743-66.
21.Wang CH, et al. J Child Neurol. 2007;22:1027-49.